Over the last years, a growing number of -omics techniques have been developed and are now widely used for genome-wide profiling of a variety of parameters and for testing specific hypotheses. A major bottleneck for such studies still is the analysis of the wealth of data that is being generated.
Our research focuses on both the development of fundamental computational and statistical methods for extracting relevant information from data (computational biology), as well as on integrating information from multiple experiments and -omics techniques to move towards a better understanding of biological systems (bioinformatics).
Our central biological interest is to decipher the molecular processes important for virus-host interaction from the cellular perspective. However, based on the fundamental approaches we have developed for that, we also moved towards working on exciting projects in immunology, cancer research and biochemistry, among others, including both basic and translational research.
Metabolic RNA labeling / Nucleotide conversion RNA sequencing
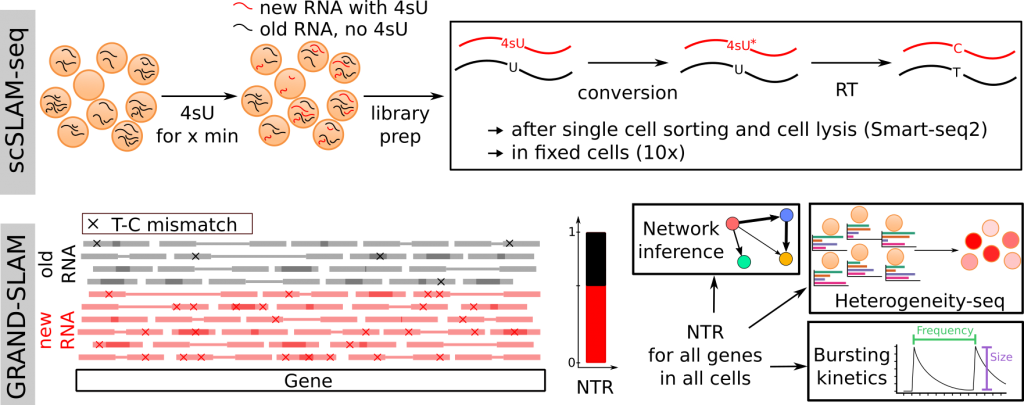
Single cell RNA sequencing (scRNA-seq) has revolutionized our view on RNA biology in individual cells. Current approaches allow to profile the total RNA levels for thousands of genes in tens or even hundreds of thousands of cells. However, scRNA-seq has one inherent limitation: Each cell can only be profiled once. This has several consequences: (i) responses to perturbations cannot be measured directly, (ii) kinetics of transcription (e.g. bursts) cannot be investigated, (iii) short-term changes due to a perturbation or stimulus within a timescale of a few hours are masked by pre-existing RNA and (iv) changes in RNA synthesis and decay cannot be differentiated. We have developed single-cell thiol(SH)-linked alkylation for the metabolic sequencing of RNA (scSLAM-seq), which integrates metabolic RNA labeling, biochemical nucleoside conversion and scRNA-seq to directly record transcriptional activity in single cells. Key to this was a new computational approach (GRAND-SLAM) that we recently developed and that allowed us to precisely quantify the new-to-total ratio (NTR) for thousands of genes in individual cells. We utilized these methods to study the earlies changes in transcription in cytomegalovirus infected fibroblasts. Subsequently, we further developed our computational tools and methods (grandR, grandRescue)
Ribo-seq
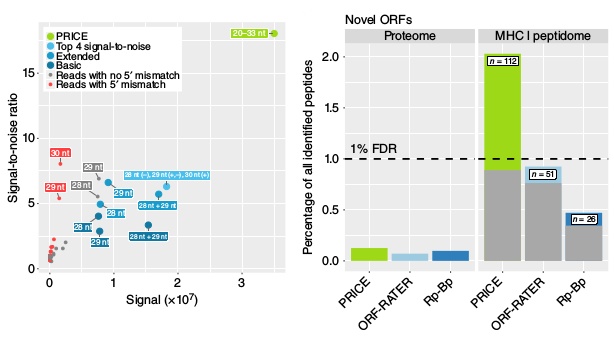
Ribosome profiling (or Ribo-seq) is a technique to identify and quantify translation of ORFs with subcodon resolution. It is based on sequencing the ribosome footprints, which are RNA fragments protected by the ribosome from enzymatic digestion. Due to stringent RNase conditions used and the matter of the fact that ribosomes translocate from codon to codon, the positions of sequencing reads show a characteristic periodicity with respect to the frame of translation. For many reads, roughly speaking, the P site codon for the corresponding ribosome is at position 12 within the read. This procedure of mapping reads to P site codons, however, is too inaccurate to properly resolve many ORFs, in particular most of the short upstream ORFs (uORFs). We have developed Probabilistic inference of codon activities by an EM algorithm (PRICE), a new algorithm to estimate P site codon positions with drastically improved signal to noise ratio. Moreover, a new statistical test included in PRICE enables us to reliable resolve complex cases such as arising with multiple overlapping ORFs. Validation of newly identified ORFs has been an unsolved issue so far. Because of them being immediately degraded after translation peptides derived from short ORFs often escape detection by mass spectrometry experiments. We reasoned that they should be well represented in MHC-I complexes, as peptide presentation via this pathway is believed to be dependent on translation rates and not on protein abundance. Indeed, we found hundreds of cryptic peptides derived from short ORFs in MHC-I ligandome experiments.
Integrative analyses & data science
Many problems in biology can only be solved by utilizing and combining more than one data set from large-scale experiments.
For instance, by integrating several data sets relevant for microRNA targeting (PAR-CLIP, Rip-Chip, LC-MS/MS, 4sU-microarrays), we have shown that viral and cellular microRNAs bind to their target sites in a context-dependent manner and that context-dependent binding has context-dependent impact on gene expression. Interestingly, we found that this context cannot be explained by the presence of absence of microRNA or target mRNA, but that other factors must be involved that constitute the context (e.g. competition with RNA binding proteins or stable RNA secondary structures).
Herpesviral genomes are relatively large (e.g. ca. 150kb for HSV-1) and are known to encode many proteins (e.g. 80 known proteins for HSV-1). Since genome sequences became available, identification of genes and proteins was based on the prediction of open reading frames (ORFs), which were then extensively validated and characterized in the 1990s. However, modern high-throughput techniques now enable a more unbiased approach to comprehensively and accurately identify genetic elements in such small genomes. By using a large array of different data sets, we were able to extend the previous annotation of HSV-1 to a total of 201 mRNAs and 284 ORFs. There were two very important lessons to learn: First, a discovery from a single large-scale data set might just represent an experimental artifact. The key for being accurate is to integrate more than one experimental technique. And second, to really understand translation and which proteins are made, the knowledge of the mRNAs (and transcription start sites) is essential.
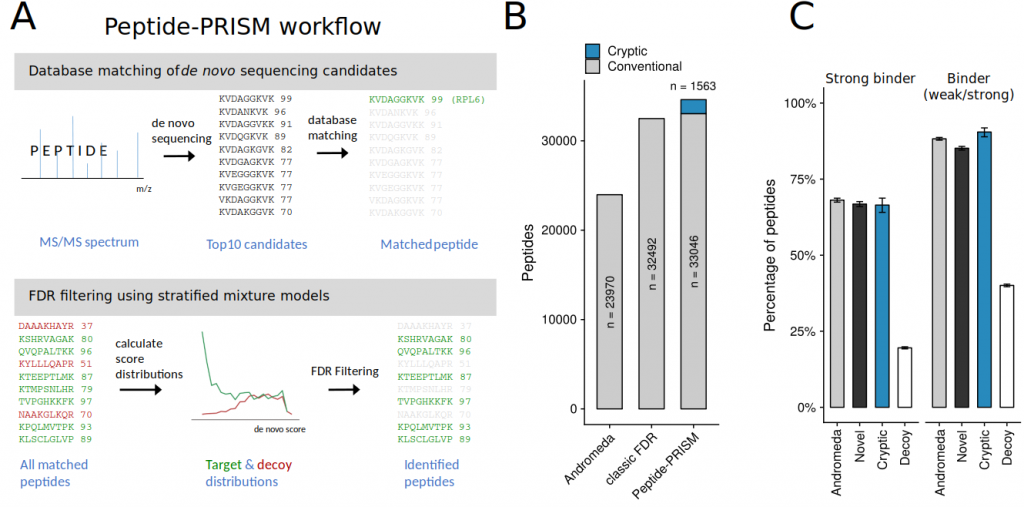
Products of short open reading frames are rapidly degraded after translation. Therefore, they have the chance to enter the MHC-I peptide presentation pathway (see above). To be able to screen the large number of immunipeptidomics data sets available, without the need to build sequence databases based on Ribo-seq data, we developed the computational approach Proteogenomic Identification using Stratified Mixture models (Peptide-PRISM). Peptide-PRISM identified thousands of cryptic peptides and showed that cryptic peptides indeed contribute up to 15% of the MHC-I ligands in different tumor samples.